什么是珠蛋白生成障碍性贫血 从三大方面深入了解它

珠蛋白生成障碍性贫血原名地中海贫血又称海洋性贫血,是一组遗传性溶血性贫血疾病。由于遗传的基因缺陷致使血红蛋白中一种或一种以上珠蛋白链合成缺如或不足所导致的贫血或病理状态。缘于基因缺陷的复杂性与多样性,使缺乏的珠蛋白链类型、数量及临床症状变异性较大。根据所缺乏的珠蛋白链种类及缺乏程度予以命名和分类。
本病广泛分布于世界许多地区,东南亚即为高发区之一。我国广东、广西、四川多见,长江以南各省区有散发病例,北方则少见。
一:病因
珠蛋白链的分子结构及合成是由基因决定的。γ、δ、ε和β珠蛋白基因组成“β基因族”,ζ和α珠蛋白组成“α基因族”。正常人自父母双方各继承2个α珠蛋白基因(αα/αα)合成足够的α珠蛋白链;自父母双方各继承1个β珠蛋白基因合成足够的β珠蛋白链。由于珠蛋白基因的缺失或点突变,肽链合成障碍导致发病。地中海贫血分为α型、β型、δβ型和δ型4种,其中以β和α地中海贫血较为常见。
1.β珠蛋白生成障碍性贫血(β地中海贫血)
β珠蛋白生成障碍性贫血(简称β地贫)的发生的分子病理相当复杂,已知有100种以上的β基因突变,主要是由于基因的点突变,少数为基因缺失。
2.α珠蛋白生成障碍性贫血(α地中海贫血)
大多数α珠蛋白生成障碍性贫血(地中海贫血)(简称α地贫)是由于α珠蛋白基因的缺失所致,少数由基因点突变造成。白基因的缺失所致,少数由基因点突变造成。
二:治疗
轻型地贫无需特殊治疗。中间型和重型地贫应采取下列一种或数种方法给予治疗。输血和去铁治疗,在目前仍是重要治疗方法之一。
1.一般治疗
注意休息和营养,积极预防感染。适当补充叶酸和维生素B12。
2.红细胞输注
输血是治疗本病的主要措施,最好输入洗涤红细胞,以避免输血反应。少量输注法仅适用于中间型α和β地贫,不主张用于重型β地贫。对于重型β地贫应从早期开始给予中、高量输血,以使患儿生长发育接近正常和防止骨骼病变。其方法是:先反复输注浓缩红细胞,使患儿血红蛋白含量达120~150g/L;然后每隔2~4周输注浓缩红细胞10~15ml/kg,使血红蛋白含量维持在90~105g/L以上。但本法容易导致含铁血黄素沉着症,故应同时给予铁螯合剂治疗。
3.铁螯合剂
常用去铁胺,可以增加铁从尿液和粪便排出,但不能阻止胃肠道对铁的吸收。通常在规则输注红细胞1年或10~20单位后进行铁负荷评估,如有铁超负荷则开始应用铁螯合剂。去铁胺,每晚1次连续皮下注射12小时,或加入等渗葡萄糖液中静滴8~12小时;每周5~7天,长期应用。或加入红细胞悬液中缓慢输注。去铁胺副作用不大,偶见过敏反应,长期使角偶可致白内障和长骨发育障碍,剂量过大可引起视力和听觉减退。维生素C与螯合剂联合应用可加强去铁胺从尿中排铁的作用。

4.脾切除
脾切除对血红蛋白H病和中间型β地贫的疗效较好,对重型β地贫效果差。脾切除可致免疫功能减弱,应在5~6岁以后施行并严格掌握适应证。
5.造血干细胞移植异基因
造血干细胞移植是目前能根治重型β地贫的方法。如有HLA相配的造血干细胞供者,应作为治疗重型β地贫的首选方法。
6.基因活化治疗
应用化学药物可增加γ基因表达或减少α基因表达,以改善β地贫的症状,已用于临床的药物有羟(经)基脲、5-氮杂胞苷(5~AZC)、阿糖胞苷、马利兰、异烟肼等,目前正在研究中。

三:预防
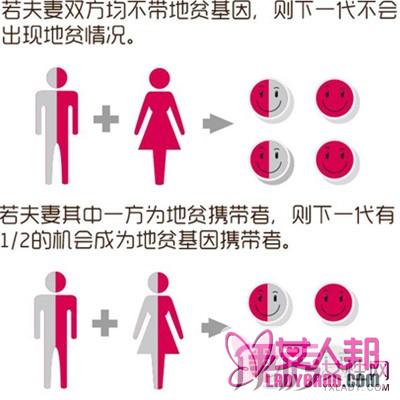
一般来说,如果两名属同一类型的地中海贫血患者结合,便有机会生下重型贫血患者。要想有效预防本病,需抽血进行肽链检测和基因分析,若证实本身和配偶同属β型极轻型或轻型地贫患者,子女将有四分之一的机会完全正常、二分之一的机会成为轻型贫血患者,四分之一的机会成为中型或重型贫血患者。鉴于本病缺少根治的方法,临床中、重型预后不良,故在婚配方面医生应向有阳性家族史或患者提出医学建议,进行婚前检查和胎儿产前基因诊断,避免下一代患儿的发生。
















{kind=link}