血友病的遗传方式是怎样的 深一步了解血友病才能预防到位

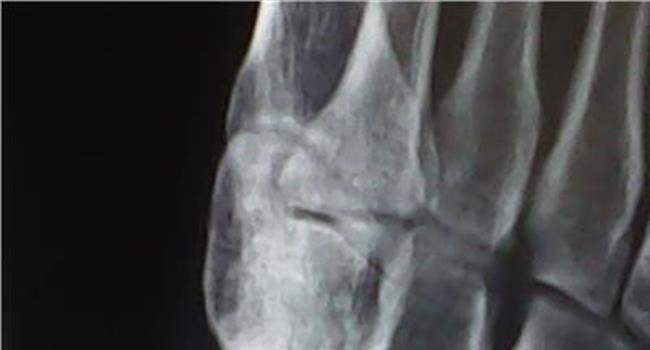
血友病为一组遗传性凝血功能障碍的出血性疾病,其共同的特征是活性凝血活酶生成障碍,凝血时间延长,终身具有轻微创伤后出血倾向,重症患者没有明显外伤也可发生“自发性”出血。
血友病甲是一种与X染色体基因有关的出血性隐性遗传病。大约每5000-10000个男性中就有一个血友病甲患者。病人因反复出现自发性出血,导致关节损伤,或畸形致残。
先天性因子Ⅷ缺乏为典型的性联隐性遗传,由女性传递,男性发病,控制因子Ⅷ凝血成分合成的基因位于X染色体。患病男性与正常女性婚配,子女中男性均正常,女性为传递者;正常男性与传递者女性婚配,子女中男性半数为患者,女性半数为传递者;患者男性与传递者女性婚配,所生男孩半数有血友病,所生女孩半数为血友病,半数为传递者。约30%无家族史,其发病可能因基因突变所致。

因子Ⅸ缺乏的遗传方式与血友病甲相同,但女性传递者中,因子Ⅸ水平较低,有出血倾向。
因子X1缺乏,均导致血液凝血活酶形成发生障碍,凝血酶原不能转变为凝血酶,纤维蛋白原也不能转变为纤维蛋白而易发生出血。
1.血友病A(血友病甲),即因子Ⅷ促凝成分(Ⅷ:C)缺乏症,也称AGH缺乏症,是一种性联隐性遗传疾病,女性传递,男性发病。
2.血友病B(血友病乙),即因子Ⅸ缺乏症,又称PTC缺乏症、凝血活酶成分缺乏症,亦为性联隐性遗传,其发病数量较血友病A少。但本型中有出血症状的女性传递者比血友病A多见。
3.血友病C(血友病丙),即因子Ⅺ(FⅪ)缺乏症,又称PTA缺乏症、凝血活酶前质缺乏症。为常染色体不完全隐性遗传,男女均可患病,是一种罕见的血友病。
15~20/10万男孩中有发病,此发病率在所调查的不同的种族和地域之间没有差异。发病率以血友病A最多占85%,血友病B占15%,血友病C较少见。

那么,简单地来说,血友病的遗传规律到底是怎样的呢?
血友病甲有四种遗传方式:
1、血友病甲患者与正常女性结婚,所生儿子为正常,女儿均为携带者。
2、正常男性与女性携带者结婚,所生儿子50%可能患有血友病甲,女儿50%可能为携带者。
3、血友病甲患者与女性携带者结婚,其女儿为血友病患者和携带者的概率各为50%,其所生儿子患病的可能性占50%。
4、男女都为血友病患者的人结婚,其所生子女均为血友病患者。
血友病乙遗传方式和血友病甲相似,血友病乙患者的女儿均为携带者,儿子为正常人。
血友病丙的遗传方式为常染色体不完全隐性遗传,男女均可患病,若临床遇到女性血友病患者,应考虑为血友病丙。
















{kind=link}