周培源数学中心 周培源应用数学研究中心
在生物体内,生物信息的流动可以分为两个部分:第一部分是存储于DNA序列中的遗传信息通过转录和翻译传入蛋白质的一级序列中,这是一维信息之间的传递,三联子密码介导了这一传递过程;第二部分是肽链经过疏水塌缩、空间盘曲、侧链聚集等折叠过程形成蛋白质的天然构象,同时获得生物活性,从而将生命信息表达出来;而蛋白质作为生命信息的表达载体,它折叠所形成的特定空间结构是其具有生物学功能的基础,也就是说,这个一维信息向三维信息的转化过程是表现生命活力所必需的。

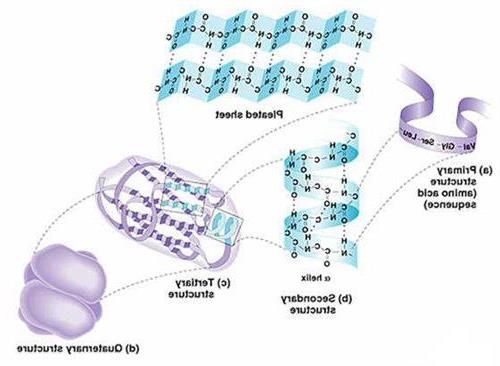
蛋白质的基本单位为氨基酸,而蛋白质的一级结构指的就是其氨基酸序列,蛋白质会由所含氨基酸残基的亲水性、疏水性、带正电、带负电……等等特性通过残基间的相互作用而折叠成一立体的三级结构。虽然蛋白质可在短时间中从一级结构折叠至立体结构,研究者却无法在短时间中从氨基酸序列计算出蛋白质结构,甚至无法得到准确的三维结构。因此,研究蛋白质折叠的过程,可以说是破译"第二遗传密码"——折叠密码(folding code)的过程。

结构决定功能,仅仅知道基因组序列并不能使我们充分了解蛋白质的功能,更无法知道它是如何工作的。蛋白质可凭借相互作用在细胞环境(特定的酸碱度、温度等)下自己组装自己,这种自我组装的过程被称为蛋白质折叠。

蛋白质折叠问题被列为"21世纪的生物物理学"的重要课题,它是分子生物学中心法则尚未解决的一个重大生物学问题。从一级序列预测蛋白质分子的三级结构并进一步预测其功能,是极富挑战性的工作。

自从20世纪60年代,Anfinsen基于还原变性的牛胰RNase在不需其他任何物质帮助下,仅通过去除变性剂和还原剂就使其恢复天然结构的实验结果,提出了"多肽链的氨基酸序列包含了形成其热力学上稳定的天然构象所必需的全部信息"的"自组装学说"以来,随着对蛋白质折叠研究的广泛开展,人们对蛋白质折叠理论有了进一步的补充和扩展。Anfinsen的"自组装热力学假说"得到了许多体外实验的证明,的确有许多蛋白在体外可进行可逆的变性和复性,尤其是一些小分子量的蛋白,但是并非所有的蛋白都如此。而且由于特殊的环境因素,体内蛋白质的折叠远非如此。

体内蛋白质的折叠往往需要有其他辅助因子的参与,并伴随有ATP的水解。因此,Ellis 于1987年提出了蛋白质折叠的"辅助性组装学说"。这表明蛋白质的折叠不仅仅是一个热力学的过程,显然也受到动力学的控制。有的学者基于有些相似氨基酸序列的蛋白质具有不同的折叠结构,而另外一些不同氨基酸序列的蛋白质在结构上却相似的现象,提出了mRNA二级结构可能作为一种遗传密码从而影响蛋白质结构的假说。但目前为止,该假说尚没有任何实验证据,只有一些纯数学论证。那么,蛋白质的氨基酸序列究竟是如何确定其空间构象的呢?围绕这一问题科研人员已进行了大量出色的工作,但迄今为止我们对蛋白质的折叠机制的认识仍是不完整的,甚至有些方面还存在着错误的观点。

自中心成立以来,国际著名应用数学家、中心名誉主任林家翘先生一直积极倡导对蛋白质折叠机理的研究,特别是对蛋白质结构的动力学过程和统计力学性质的研究。中心先后邀请国内外相关领域许多著名学者到中心访问、讲学,并与中心科研人员展开了广泛而深入的交流合作。

从2006年开始,中心与美国麻省理工学院的著名统计物理学家黄克孙先生合作提出研究蛋白质折叠的"CSAW" 模型,实现了对中小型蛋白质结构的快速折叠;近期进一步开展了对蛋白质折叠统计热力学原理的研究。通过研究原子、分子间相互作用,掌握蛋白质系综内部微观状态的结构特征、能量分布规律,以期从微观角度解释宏观可观测物质属性。

蛋白质折叠理论工作获得了清华大学骨干人才支持计划研究(2007)"基于条件自回避行走的蛋白质折叠机理研究"和清华大学自主科研计划(2010)"蛋白质折叠的统计热力学原理"的支持,极大地推动了中心在蛋白质科学方面的研究进展。

周培源应用数学研究中心热忱欢迎国内外研究学者的访问和交流合作,积极推动中心在蛋白质科学研究领域的不断进步。
















{kind=link}